Abstract
Huntington’s disease (HD) is a fatal neurodegenerative disorder caused by a polyglutamine expansion in the huntingtin (HTT) protein. The expression of mutant HTT in the baker’s yeast Saccharomyces cerevisiae recapitulates many of the cellular phenotypes observed in mammalian HD models. Mutant HTT aggregation and toxicity in yeast is influenced by the presence of the Rnq1p and Sup35p prions, as well as other glutamine/asparagine-rich aggregation-prone proteins. Here we investigated the ability of a subset of these proteins to modulate mutant HTT aggregation and to substitute for the prion form of Rnq1p. We find that overexpression of either the putative prion Ybr016wp or the Sup35p prion restores aggregation of mutant HTT in yeast cells lacking the Rnq1p prion. These results indicate that an interchangeable suite of aggregation-prone proteins regulates mutant HTT aggregation dynamics in yeast, which may have implications for mutant HTT aggregation in human cells.
Funding Statement
Flaviano Giorgini was supported by a MRC New Investigator award (G0700090). The authors have declared that no competing interests exist.Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease 1,2 caused by the expansion of a polyglutamine (polyQ) tract in the huntingtin (HTT) protein, which leads to its misfolding and aggregation3 . Intracellular HTT aggregates are a pathological hallmark of HD4,5, with their morphology and localization influencing downstream cellular effects6,7 . Several studies suggest that macromolecular HTT aggregates which prevent further intermolecular interactions are neuroprotective8, while soluble oligomeric species of mutant HTT capable of sequestering other cellular proteins are neurotoxic9 . HD has been modelled in a wide range of organisms10,11 including the baker’s yeast, Saccharomyces cerevisiae12. Yeast models recapitulate many of the cellular disease phenotypes seen in HD patients, including disturbances in mitochondrial function, increased production of reactive oxygen species, disruption of the cytoskeleton, proteasome dysfunction and increased flux through the kynurenine pathway of tryptophan degradation13 .
A number of yeast models have been developed to study the cellular effects of mutant HTT expression7,12,14 . These models have shown that aggregation and cellular toxicity of mutant HTT in yeast is dependent upon the presence of the Rnq1p in its prion [PIN+] conformation14 , as deletion of RNQ1 or curing of [PIN+] ameliorates these phenotypes. Furthermore, a network of prion-like glutamine-rich proteins modulates mutant HTT-dependent toxicity in yeast15 , with expression of these proteins – as well as Rnq1p – having an additive effect on the overall level of toxicity observed. Although the cellular function of Rnq1p is unclear, it is crucial for the de novo formation of [PSI+], the prion form of Sup35p16,17 . [PSI+] is one of the most extensively studied yeast prions and is encoded by SUP35, with the non-prion form of the protein being crucial for the release of newly synthesized polypeptides from the ribosomal complex through ATP hydrolysis18 . A recent study found that Sup35p interacts with mutant HTT through its Q/N-rich prion domain, and the presence of [PSI+] is critical for full toxicity of mutant HTT constructs with extended polyproline regions19 .
We previously identified a number of putative yeast prions in a screen for gene deletion suppressors of HTT103Q-dependent toxicity in yeast20 . The proteins in question – Def1p, Ybr016wp, Yir003wp and Ylr278cp – all have glutamine/asparagine-rich (Q/N-rich) domains and computational analyses suggests that they share primary sequence features with known prions21 . Subsequent work has found that Def1p, Yir003wp and Ylr278cp do not behave as typical yeast prion proteins, while Ybr016wp has a large number of characteristics in common with established yeast prions such as Sup35p22 . Here we further investigate the effect of Ybr016wp and Sup35p on mutant HTT aggregation dynamics in yeast. In order to unmask the effect these proteins have on this protein misfolding process, we focused our attention on changes in HTT aggregation dynamics in a rnq1∆ background. We find that both Sup35p and Ybr016wp modulate the formation of mutant HTT aggregates in yeast and that these effects are partially dependent on the presence of Rnq1p in the cell. In total, this work indicates that other aggregation-prone proteins can substitute for Rnq1p in the context of HTT misfolding, and suggests that dynamic interplay between a suite of such proteins modulates HTT aggregation in yeast, which may have implications for HTT aggregation in human cells.
Methods
Yeast strains and culturing: The BY4741 and rnq1Δ deletion strains (MAT a,
Yeast growth assays: Yeast colonies were inoculated into 100 µl of medium in 96-well plates and incubated at 30 ºC for 16 to 18 hours. Cultures were serially diluted by 5 or 10-fold in distilled water and spotted (5 μl) onto selective plates containing either 2 % galactose or 2 % glucose as a carbon source and incubated for 60 to 72 hours at 30 ºC.
Fluorescence microscopy: Colonies were inoculated in 5 ml of glucose containing selective medium and incubated at 30 ºC with shaking for 24 hours. Cultures were washed once with water and diluted to an OD600 of 0.2 in 5 ml selective media containing 2 % raffinose. After a further 6 hours at 30 ºC with shaking, galactose was added to a final concentration of 2 % to induce expression of constructs under the control of GAL1 promoters. After a further 12 hours the number of cells containing aggregates was scored using a Zeiss Axioscope 2 fluorescent microscope with a Zeiss 100 X Plan-NEOFLUAR (1.30/oil, ∞/0.17) objective and a chromomycin filter.
Determination of yeast [PIN] and [PSI] status: The presence of the [PIN+] prion was detected by a previously described protocol24 using a primary antibody against Rnq1p (Santa Cruz Biotechnology Inc., 1:10,000 dilution). Samples for the determination of [PSI] status were grown the same way as for the [PIN] status assay, and 4 OD units harvested by centrifugation and suspended in 100 µl of ice-cold lysis buffer [1X PBS without Ca2+and Mg2+(PAA Laboratories GmbH, Austria), 100 mM NaCl, 2 mM PMSF and 1x EDTA-free protease inhibitors (Roche)]. Samples were lysed with 100 µl of acid washed glass beads (425-600 μm, Sigma-Aldrich, USA) in a bead-beater for 1 minute at maximum speed. After adding an additional 100 µl of ice-cold lysis buffer, samples were left on ice for 1 minute to allow the beads to sediment and the supernatant was removed for further analysis. The amount of protein was quantified using a NanoPhotometer™ Pearl (IMPLEN GmbH, Germany) and 10 μg of protein in 50 μl of lysis buffer were spun at 50,000 rpm at 4 ºC for 15 minutes in a BECKMAN TL-100 ultracentrifuge (Beckman, USA). The supernatant was removed and used as the “Soluble” fraction, while the pellet was resuspended in 50 μl of lysis buffer and used as the “Pellet” fraction. Protein from the total, soluble and pellet fractions were mixed with 2 µl of 5 X protein loading dye [50 mM Tris-HCl (pH 6.8), 2 % SDS, 10 % glycerol, 1 % β-mercaptoethanol, 12.5 mM EDTA, and 0.02 % bromophenol blue] to give a final volume of 10 μl. Samples were denatured at 95 ºC for 5 minutes, separated by SDS-PAGE, and transferred to PVDF membranes. Sup35p was detected with a primary antibody against yeast Sup35p (1:10,000 dilution), a generous gift from Mick Tuite (University of Kent, UK).
Statistical Analyses: Data was analysed by unpaired, two-tailed Mann-Whitney tests using Prism 6 (GraphPad Software).
Results
HD model yeast exhibit HTT aggregation independent of toxicity in a [PIN+] and [PSI+] background: To avoid confounding issues due to variable plasmid copy number we generated yeast strains containing either HTT25Q or HTT103Q constructs integrated at the HIS3 locus in parental BY4741 yeast, as well as in a rnq1Δ strain in the same background. The Rnq1p-deficient strain was generated by deleting RNQ1 using homologous recombination, which was verified by PCR genotyping and immunoblotting (Figure 1b; data not shown). These constructs encode the first 17 amino acids of HTT followed by the respective polyQ length under the control of an inducible GAL1 promoter, and have an N-terminal FLAG epitope tag and a C-terminal CFP fusion. Integration of the HTT constructs was achieved by employing the integrative vector pRS303, and successful integration events were confirmed by PCR and sequencing of the HIS3 locus (data not shown).
a) Expression of the integrated HTT103Q construct in BY4741 yielded no toxicity or cell death, even when Rnq1p was expressed on a high copy number plasmid, labelled here as 2µ. Equal numbers of cells were serially diluted threefold and plated on medium containing glucose to assess cell numbers and medium containing galactose to induce the expression of mutant HTT. b) [PIN+] is present in the integrated BY4741 strains and is absent in the rnq1Δ background. (T=total protein, S=soluble protein, and P=insoluble protein). The [PIN] status of the cells was determined by using standard immunoblot techniques and an antibody raised against yeast Rnq1p. c) [PSI+] is present in both the integrated BY4741 strains and the rnq1Δ background. The presence of [PSI+] was assessed using an antibody raised against yeast Sup35p.
Fig. 1: An integrated HD yeast model exhibits a reduced level of mutant HTT toxicity in the cell independent of the presence of [PIN+] and [PSI+]
The effect of expressing the integrated HTT constructs was assessed via growth assays and fluorescence microscopy. Surprisingly, we observed that expression of HTT103Q in the integrated strain did not impair growth in multiple integrants generated from independent transformations (Figure 1a). As the presence of the [PIN+] and [PSI+] prions modulates mutant HTT aggregation and toxicity in baker’s yeast25 , we assessed their status in the integrated strains. We observed that Rnq1p is predominantly found as [PIN+] in the BY4741 integrated strains, and as expected is not present in the rnq1Δ strains (Figure 1b). Sup35p (Figure 1c) was found as [PSI+] in both the BY4741 and rnq1Δ integrated strains. These data confirm that lack of [PIN+] or [PSI+] is not responsible for the absence of HTT103Q toxicity in the generated strains.
Despite the lack of HTT103Q-dependent toxicity in the newly created integrated strains (Figure 1a), we observed a high level of inclusion body formation in these cells (Figure 2). Indeed, ~90 % of the cells in the integrated HTT103Q strain contained mutant HTT inclusion bodies 12 hours post induction (Figure 2), while in the rnq1Δ strain the number of cells containing inclusions is reduced to ~7 %. Reintroduction of a plasmid expressing Rnq1p into the rnq1Δ strain restored inclusion body formation to control levels. Thus, this novel integrated yeast model of HD dissociates the toxicity caused by HTT103Q expression from its aggregation and allowed us to further dissect the relationship between prion-like yeast proteins and the dynamics of mutant HTT aggregation without the confounding effects of toxicity.
Expression of HTT was induced in overnight cultures and the number of cells containing inclusion bodies was assessed by fluorescence microscopy. Overexpression of Rnq1p in a rnq1Δ background restores inclusion body formation in cells expressing HTT103Q. Counts were converted to the percentage of cells containing inclusion bodies and statistical analyses were performed using unpaired, two-way Mann-Whitney tests (**P< 0.01, ***P< 0.001), NS = not significant. All data are shown as the mean ± SEM; n ≥ 5 per genotype.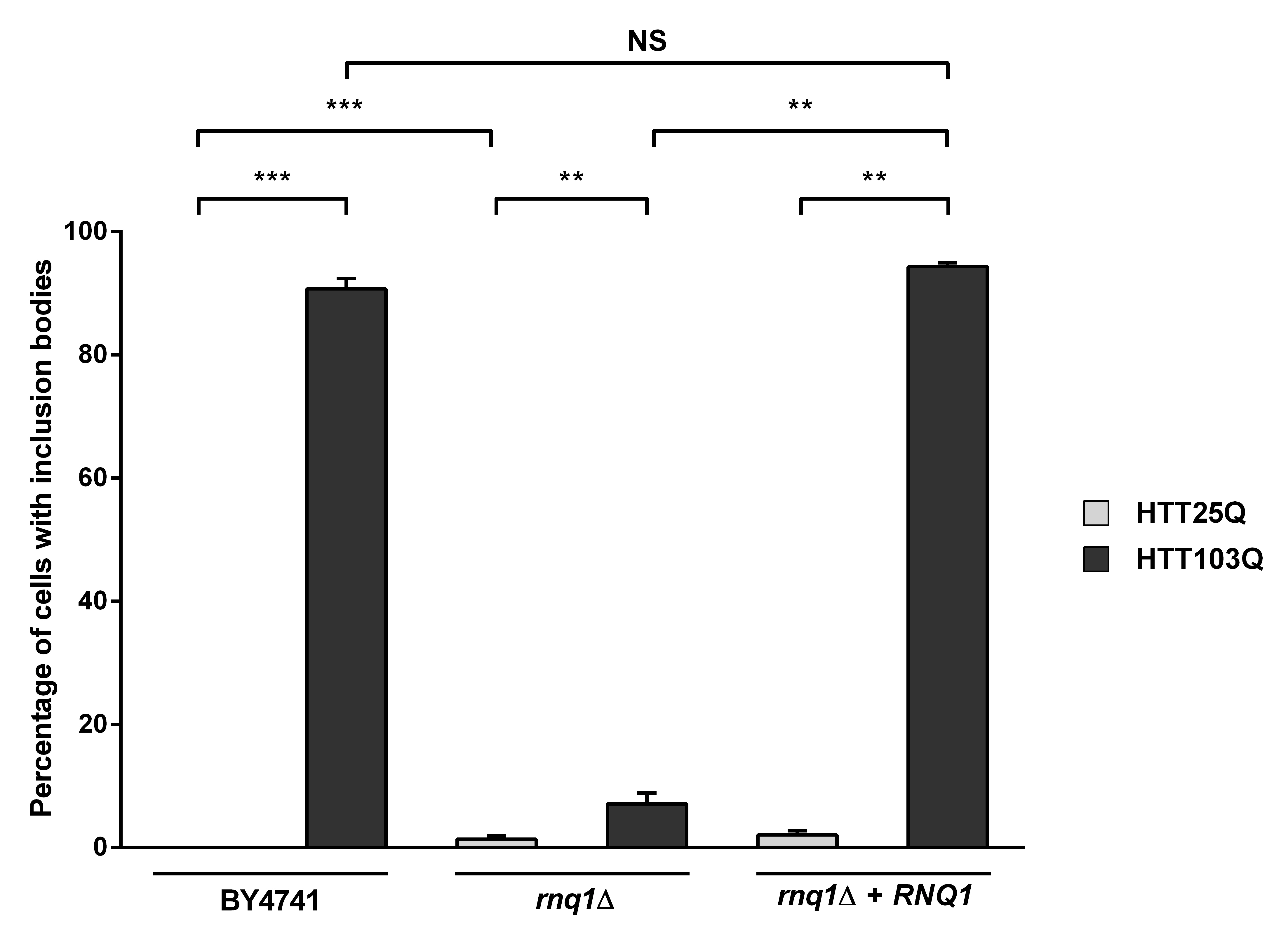
Fig. 2: Overexpression of Rnq1p increases the aggregation of mutant huntingtin in yeast
Interestingly, we found that deletion of the endogenous RNQ1 gene leads to the formation of inclusion bodies in a small number of cells expressing the HTT25Q construct, which does not normally form these structures (Figure 2; P=0.0002). It has recently been observed that a network of proteins in yeast modify the formation of mutant HTT aggregates26 , and it is thus possible that deletion of the endogenous RNQ1 disrupts the balance between other yeast genes that govern HTT aggregation, leading to the aggregation of the HTT25Q construct, which is highly enriched for glutamines in comparison to most endogenous yeast proteins.
Ybr016wp and Sup35p modulate HTT aggregation independently of Rnq1p: We next focused on characterizing the role of Ybr016wp and Sup35p in the aggregation dynamics of mutant HTT in yeast. We initially examined the ability of Ybr016wp to modulate the number of cells containing inclusion bodies when overexpressed in the presence and absence of Rnq1p (Figure 3). Interestingly, overexpression of Ybr016wp causes a small but significant increase in the formation of inclusion bodies in HTT25Q-expressing BY4741 (~ 12 %; P=0.0022) and rnq1Δ (~ 7 %; P=0.0007) cells (Figure 3). A possible explanation for this is the high level of Ybr016wp overexpression, which contains five consecutive perfect repeats (GYNQQ) that are enriched for asparagine, glutamine, tyrosine and glycine. Overexpression of such sequences induces aggregation of other proteins, and could seed formation of inclusion bodies by polyQ-rich sequences22,27 . Analysis of the HTT103Q integrated strain revealed that Ybr016wp overexpression yileds a small but significant increase in the number of BY4741 cells exhibiting inclusion bodies (P=0.008). Ybr016wp overexpression in the rnq1Δ background also significantly increased the number of rnq1Δ cells forming HTT103Q inclusions, from ~ 7 % to ~ 16 % (P=0.0293). These data indicate that Ybr016wp is able to partially compensate for the loss of Rnq1p in the rnq1Δ background with respect to mutant HTT aggregation, and that this aggregation is not polyQ-length dependent.
Expression of HTT was induced in overnight cultures and the number of cells containing inclusion bodies was assessed by fluorescence microscopy. Overexpression of Ybr016wp in arnq1Δ background increases inclusion body formation in both HTT25Q and HTT103Q expressing cells. Counts were converted to the percentage of cells containing inclusion bodies and statistical analyses were performed using unpaired, two-way Mann-Whitney tests (*P< 0.05, **P< 0.01, ***P< 0.001). All data are shown as the mean ± SEM; n ≥ 6 per genotype.
Fig. 3: Overexpression of Ybr016wp increases the aggregation of mutant huntingtin in yeast
As the presence of Rnq1p is crucial for the de novo formation of [PSI+] as well as the formation of mutant HTT aggregates in yeast, we next investigated the connection between Sup35p expression and mutant HTT aggregation by overexpressing Sup35p. As with Ybr016wp, overexpression of Sup35p leads to a small but significant increase in the formation of HTT25Q inclusion bodies in the BY4741 background (Figure 4; P < 0.0001). It is possible that these inclusion bodies form due to the presence of [PSI+] in the cells, as formation of [PSI+] can be triggered by overexpression of Sup35p28 , which is able to cause aggregation of heterogeneous polyQ-rich sequences29 . Interestingly, in the rnq1Δ strain overexpressing Sup35p the formation of HTT25Q inclusion bodies is dramatically increased (~ 68 %) compared with the parental BY4741 strain (~ 12 %; P=0.0264). As mentioned earlier a network of proteins is able to modify aggregation of mutant HTT in yeast25 and has also been found to trigger the formation of [PSI+]. It is therefore possible that this network is activated by the absence of endogenous Rnq1p in the rnq1Δ strain, and is further stimulated by the presence of [PSI+]. Interestingly, the HTT25Q and HTT103Q strains exhibit a similar number of cells with aggregates in the rnq1Δ background with Sup35p expression, indicating that this substitution is not polyQ-length dependent, and potentiates the aggregation of shorter polyQ stretches.
Expression of HTT was induced in overnight cultures and the number of cells containing inclusion bodies was assessed by fluorescence microscopy. Overexpression of Sup35p in a rnq1Δ background greatly enhances inclusion body formation in a non-polyglutamine dependent manner. Counts were converted to the percentage of cells containing inclusion bodies and statistical analyses were performed using unpaired, two-way Mann-Whitney tests (*** P< 0.001, ****P< 0.0001), NS = not significant. All data are shown as the mean ± SEM; n ≥ 7 per genotype.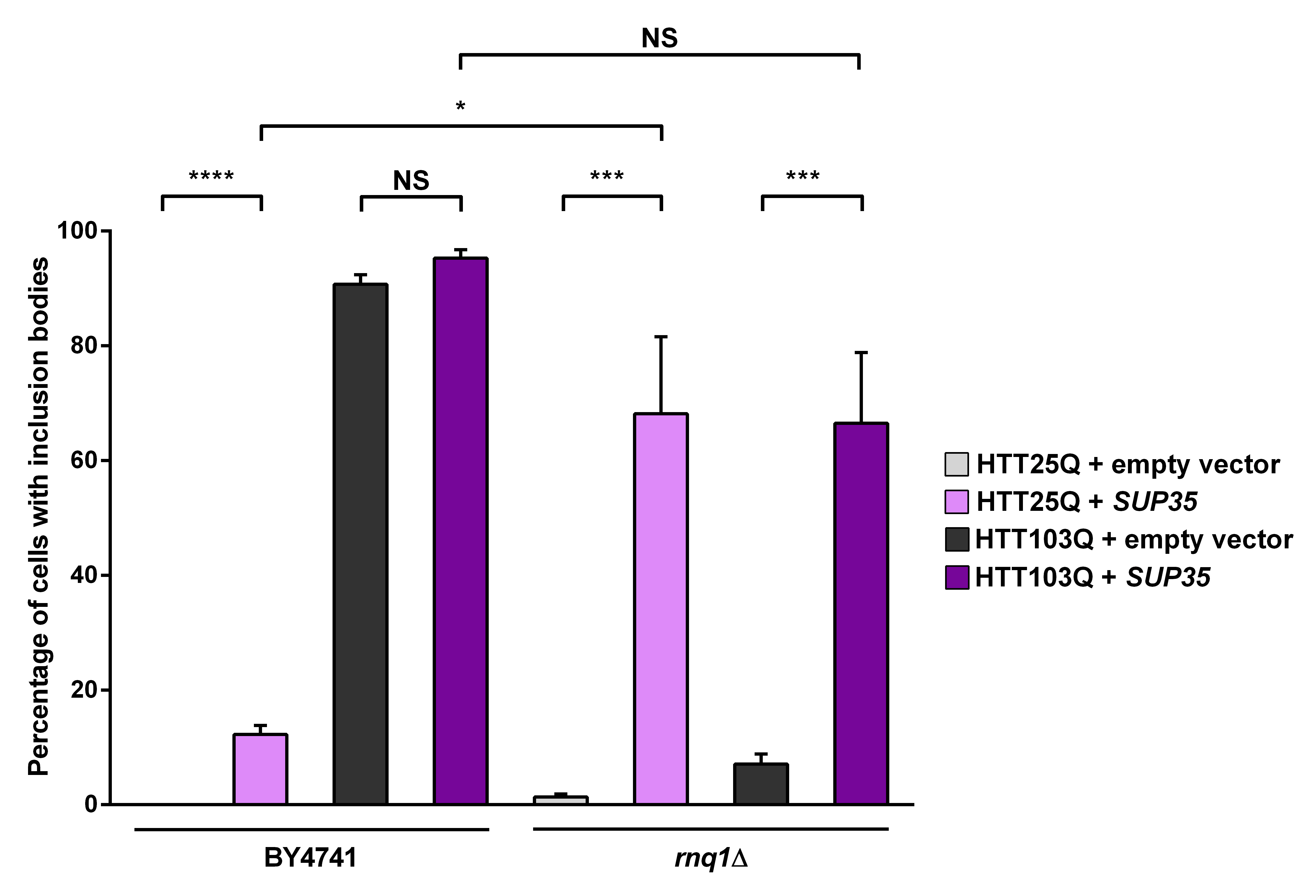
Fig. 4: Overexpression of Sup35p increases the aggregation of mutant huntingtin in yeast
Discussion
Our studies indicate that overexpression of Rnq1p, Sup35p and Ybr016wp can modulate the formation of mutant HTT aggregates in yeast. Furthermore, the induction of mutant HTT inclusion body formation by both Sup35p and Ybr016wp is in part dependent upon the presence of Rnq1p in the cell. The mechanism(s) by which these two aggregation-prone proteins modulate mutant HTT aggregation is not clear, nor is it known if they are acting in a similar manner. In the case of Sup35p it has been proposed that there are two groups of factors that govern both the formation of mutant HTT aggregates and the Sup35p to [PSI+] switch in yeast26 . The first group is involved in the formation of large mutant HTT aggregates and the switch between Sup35p and [PSI+], while the factors in the second group are crucial for the formation of soluble oligomeric species of mutant HTT, as well as the formation of diffuse [PSI+] aggregates. These findings further support the hypothesis that yeast prions and glutamine-rich amyloidogenic proteins interact with similar molecular partners upon forming larger amyloid structures in yeast22 . Based upon this observation it is possible that mutant HTT interacts directly with Sup35p. Indeed, a recent investigation indicates that mutant HTT can sequester Sup35p in [PSI+] cells27 . As most established yeast prions have a Q/N-rich sequence and mutant HTT can sequester Q-rich sequences in yeast28 , it is possible that mutant HTT can sequester other yeast prions. Ybr016wp is poorly characterised, and the relationship between this protein and mutant HTT described here is novel. Although Ybr016wp has an unknown cellular function it contains a CYSTM domain that is characteristic of eukaryotic proteins involved in stress tolerance30 and is anchored to the plasma membrane and predicted to be palmitoylated, similar to the human prion protein PrPC 31.
Many highly conserved yeast proteins have prion-like features22 , suggesting that human proteins with prion-like domains may behave in a similar manner. Indeed, a recent study has implicated a large number of glutamine and asparagine rich human RNA-binding proteins in the pathology of a range of neurodegenerative diseases32. Thus, it is possible that a suite of aggregation prone proteins that modify the aggregation of mutant HTT exists not only in yeast but in humans as well, which may have implications for HD pathogenesis. Indeed, such proteins could modify the aggregation and misfolding of not only mutant HTT, but other amyloidogenic proteins as well, thus informing the development of therapies for HD as well as for other neurodegenerative diseases caused by protein misfolding.
Acknowledgements
We thank Susan Lindquist (Whitehead Institute, USA) for providing the pRS303Htt integrative plasmids. We are grateful to Mick Tuite (University of Kent, UK) for the Sup35 antibody. We acknowledge Gary Jones (NUI Maynooth, Ireland) for his helpful comments and advice.References
- Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet. 2003 May 10;361(9369):1642-4. PubMed PMID:12747895.
- Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011 Jan;10(1):83-98. PubMed PMID:21163446.
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993 Mar 26;72(6):971-83. PubMed PMID:8458085.
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997 Sep 26;277(5334):1990-3. PubMed PMID:9302293.
- Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998 Jun 1;141(5):1097-105. PubMed PMID:9606203.
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004 Oct 14;431(7010):805-10. PubMed PMID:15483602.
- Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci U S A. 2006 Jul 18;103(29):11045-50. PubMed PMID:16832050.
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007 Aug 9;448(7154):704-8. PubMed PMID:17687326.
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004 Jun;36(6):585-95. PubMed PMID:15146184.
- Parker JA, Connolly JB, Wellington C, Hayden M, Dausset J, Neri C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci U S A. 2001 Nov 6;98(23):13318-23. PubMed PMID:11687635.
- Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005 Oct 5;25(40):9152-61. PubMed PMID:16207874.
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000 Feb 15;97(4):1589-94. PubMed PMID:10677504.
- Mason RP, Giorgini F. Modeling Huntington disease in yeast: perspectives and future directions. Prion. 2011 Oct-Dec;5(4):269-76. PubMed PMID:22052350.
- Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002 Jun 10;157(6):997-1004. PubMed PMID:12058016.
- Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proc Natl Acad Sci U S A. 2006 Jul 18;103(29):11051-6. PubMed PMID:16832049.
- Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997 Oct;147(2):507-19. PubMed PMID:9335589.
- Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell. 2001 Jul 27;106(2):171-82. PubMed PMID:11511345.
- Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005 Jun;6(6):435-50. PubMed PMID:15931169.
- Gong H, Romanova NV, Allen KD, Chandramowlishwaran P, Gokhale K, Newnam GP, Mieczkowski P, Sherman MY, Chernoff YO. Polyglutamine toxicity is controlled by prion composition and gene dosage in yeast. PLoS Genet. 2012;8(4):e1002634. PubMed PMID:22536159.
- Giorgini F, Guidetti P, Nguyen Q, Bennett SC, Muchowski PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat Genet. 2005 May;37(5):526-31. PubMed PMID:15806102.
- Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A. 2000 Oct 24;97(22):11910-5. PubMed PMID:11050225.
- Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009 Apr 3;137(1):146-58. PubMed PMID:19345193.
- Sherman, F., Fink, G. R. & Hicks, J. B. (1986). Methods in Yeast Genetics.., New York: Cold Spring Harbor Laboratory Press
- Tauber E, Miller-Fleming L, Mason RP, Kwan W, Clapp J, Butler NJ, Outeiro TF, Muchowski PJ, Giorgini F. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J Biol Chem. 2011 Jan 7;286(1):410-9. PubMed PMID:21044956.
- Zhao X, Park YN, Todor H, Moomau C, Masison D, Eisenberg E, Greene LE. Sequestration of Sup35 by aggregates of huntingtin fragments causes toxicity of [PSI+] yeast. J Biol Chem. 2012 Jul 6;287(28):23346-55. PubMed PMID:22573320.
- Manogaran AL, Hong JY, Hufana J, Tyedmers J, Lindquist S, Liebman SW. Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS Genet. 2011 May;7(5):e1001386. PubMed PMID:21625618.
- Toombs JA, McCarty BR, Ross ED. Compositional determinants of prion formation in yeast. Mol Cell Biol. 2010 Jan;30(1):319-32. PubMed PMID:19884345.
- Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997 May 30;89(5):811-9. PubMed PMID:9182769.
- Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci U S A. 2004 Aug 31;101(35):12934-9. PubMed PMID:15326312.
- Venancio TM, Aravind L. CYSTM, a novel cysteine-rich transmembrane module with a role in stress tolerance across eukaryotes. Bioinformatics. 2010 Jan 15;26(2):149-52. PubMed PMID:19933165.
- Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel. 2008 Nov;21(11):639-44. PubMed PMID:18753194.
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012 Jun 26;1462:61-80. PubMed PMID:22445064.
Leave a Comment
You must be logged in to post a comment.