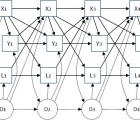
Insufficient evidence exists to guide the long-term pharmacological management of Huntington’s disease (HD) although most current interventions rely on symptomatic management. The effect of many frontline treatments on potential endpoints for HD clinical trials remains unknown. Our objective was to investigate how therapies widely used to manage HD affect the symptom for which they are prescribed and other endpoints using data from TRACK-HD. We used longitudinal models to estimate effects of medication use on performance on tests of motor, cognitive and neuropsychiatric function using data from 123 TRACK-HD stage 1/2 participants across four study visits. Adjustment for confounding by prior medication use, prior clinical performance, concomitant use of other medications, and baseline variables (sex, disease group, age, CAG, study site, education) enabled a closer-to-causal interpretation of the associations. Adjusting for baseline variables only, medication use was typically associated with worse clinical performance, reflecting greater medication use in more advanced patients. After additional adjustment for longitudinal confounders such “inverse” associations were generally eliminated and in the expected directions: participants taking neuroleptics tended to have better motor performance, improved affect and poorer cognitive performance, and those taking SSRI/SNRIs had less apathy, less affect and better total behaviour scores. However, we uncovered few statistically significant associations. Limitations include sample size and unmeasured confounding. In conclusion, adjustment for confounding by prior measurements largely eliminated associations between medication use and poorer clinical performance from simple analyses. However, there was little convincing evidence of causal effects of medication on clinical performance and larger cohorts or trials are needed.