Abstract
Since the ever-increasing availability of phylogenetic informative data, the last decade has seen an upsurge of ecological studies incorporating information on evolutionary relationships among species. However, detailed species-level phylogenies are still lacking for many large groups and regions, which are necessary for comprehensive large-scale eco-phylogenetic analyses. Here, we provide a dataset of 100 dated phylogenetic trees for all European tetrapods based on a mixture of supermatrix and supertree approaches. Phylogenetic inference was performed separately for each of the main Tetrapoda groups of Europe except mammals (i.e. amphibians, birds, squamates and turtles) by means of maximum likelihood (ML) analyses of supermatrix applying a tree constraint at the family (amphibians and squamates) or order (birds and turtles) levels based on consensus knowledge. For each group, we inferred 100 ML trees to be able to provide a phylogenetic dataset that accounts for phylogenetic uncertainty, and assessed node support with bootstrap analyses. Each tree was dated using penalized-likelihood and fossil calibration. The trees obtained were well-supported by existing knowledge and previous phylogenetic studies. For mammals, we modified the most complete supertree dataset available on the literature to include a recent update of the Carnivora clade. As a final step, we merged the phylogenetic trees of all groups to obtain a set of 100 phylogenetic trees for all European Tetrapoda species for which data was available (91%). We provide this phylogenetic dataset (100 chronograms) for the purpose of comparative analyses, macro-ecological or community ecology studies aiming to incorporate phylogenetic information while accounting for phylogenetic uncertainty.
Funding Statement
The research leading to this paper had received funding from the European Research Council under the European Community’s Seven Framework Programme FP7/2007–2013 Grant Agreement no. 281422 (TEEMBIO). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.Introduction
The use of phylogenetic data into ecological analyses has grown rapidly in the last decades, giving rise to new disciplines such as community phylogenetics which incorporate information on species relatedness into the study of community structure1,2, as well as to studies of large-scale distribution of species and their phylogenetic diversity3,4. Additionally, the integration of ecological and evolutionary information holds promise to improve ecological forecasting in the current context of climate and land change and biodiversity loss5,6. Since the pioneering work of Dan Faith7, conservation biology has long recognized the importance of considering phylogenetic diversity as a relevant feature for conservation8,9. The EDGE framework is, in this regard, an important initiative that combines the evolutionary distinctiveness of species (i.e. the evolutionary contribution of a species to the tree of life) with globally endangered risk assessment to derive conservation priorities10. Recent works have also focused on how future climate and land use change could further jeopardize the tree of life in certain parts of the world11,12.
To foster the developments of these emergent fields and timely questions, detailed and broadly sampled phylogenetic hypotheses are needed to appropriately integrate evolutionary information into ecological and conservation studies. Recent phylogenomic studies have improved our understanding of the evolutionary relationships within the main Tetrapoda groups, especially at high levels such as families and orders. For instance, Roelants and colleagues13 clarified the relationships between global amphibians at the family level, while Pyron et al.14 performed a similar achievement on Squamata, sampling all families and sub-families. Concerning birds, Hackett et al.15 elucidated the inter-ordinal relationships of extant birds, and a later study16 confirmed the partly controversial results found by Hackett and colleagues. Despite these achievements, we still lack detailed species-level phylogenies for such groups. Moreover, there is a lack of phylogenies for particular regions (but see 17), as systematists mainly focus on building species-level phylogenies for entire clades. Although it is of obvious interest, research areas such as community phylogenetics and conservation planning do not specifically require complete taxonomic sampling, but rather complete spatial, or biogeographic, sampling. In order words, ecological studies that wish to integrate evolutionary data usually require a phylogenetic hypothesis for the entire species pool under study, which might be along a specific gradient18 or a continental scale assessment11,19 . For instance, incorporating phylogenetic diversity in reserve design or gap analysis only require a complete phylogenetic tree for the entire group with the region of interest (see for example 19, 85). It should however be noted that since the complete coverage only concerns Europe, estimates of phylogenetic uniqueness are therefore biased and should be accounted for in the analysis of the data (e.g. 86).
For that purpose, we here construct and provide a phylogenetic dataset for all Tetrapoda species that occur in the entire European sub-continent (including Turkey) built on relevant phylogenetic data in Genbank and consensus tree knowledge, based on a supermatrix-supertree mixed approach20 . We also check the congruence of the phylogenies obtained with previous evolutionary studies.
Methods
Squamata and Testudinae
The list of European Squamata species was extracted from Maiorano et al21. DNA sequences of 7 nuclear (BDNF, c-mos, NT3, PDC, R35, RAG-1, RAG-2) and 6 mitochondrial loci (12S, 16S, COI, cytB, ND2, ND4) were downloaded from Genbank with PHLAWD22. These regions have been shown to be useful for phylogenetic inference in previous studies of squamates according to Pyron et al14. Only 16 species of a total of 239 had no molecular data available in Genbank. In addition to Squamata species, we included 3 levels of outgroup taxa: Sphenodon punctata (closest living relative to Squamata); all 10 species of European turtles, two crocodilians (Alligator and Crocodylus) and two birds (Dromaius and Gallus); and finally two mammals (Mus and Pan). Genbank accession numbers are detailed in Table S1 (Appendix 1).
For each region, DNA sequences were aligned with MAFFT23 and checked by eye with Seaview24. Ambiguous alignment positions were trimmed with trimAl25. All the regions were concatenated in a supermatrix with FASConCAT26. The phylogenetic inference analysis was conducted with RaxML v. 7.8.127 using the GTRGAMMA model and employing the rapid hill-climbing algorithm28; we searched for 100 Maximum Likelihood trees applying a family tree constraint for squamates based on Pyron et al14. Bootstrapping was conducted with 1000 replicates to assess clade support.
The 100 ML trees were dated with penalized-likelihood as implemented in r8s29; we constrained 5 nodes based on fossil information extracted from Mulcahy et al.30: we set a minimum and a maximum age of 256 and 300 mya respectively for the most recent common ancestor (mrca) of all Reptilia31,32, a minimum and a maximum age of 239 and 250 mya respectively for the mrca of Birds and crocodilians32 , a minimum age for the mrca of Lepidosauria of 223 mya33,34, a minimum age of 111 mya for the stem branch of Amphisbaenidae34,35, and a minimum age of 93 mya for the stem branch of Alethinophidia36. The best smoothing value was determined by a cross-validation procedure, following 29 .
The data matrix and the phylogenetic tree with the highest likelihood are available in Treebase (accession number: S15708).
Amphibians
For Amphibians, we include here the phylogenetic tree constructed for a previous study19. The list of European Amphibian species was extracted from Maiorano et al21. We retrieved from GenBank sequences of phylogenetic informative regions that were available for at least 30% of the species: 9 mitochondrial (12S, 16S, COI, cytb, ND1, ND2, ND4, tRNA-Leu, tRNA-Val) and 2 nuclear (RAG-1, rho) regions. We found relevant molecular data for all species, but we excluded the two hybrid species Pelophylax grafi and Pelophylax hispanicus. We included Xenopeltis unicolor, Gallus gallus and Mus musculus as outgroups to root the tree. For each region, alignment was conducted with four programs (Clustal37, Kalign
We dated the 100 ML trees with penalized-likelihood (r8s) using the following fossil data to constrain minimum ages for selected nodes: 155 mya for the crown-origin of salamanders40, 170 mya for Bombianura41, 250 mya for Batrachia42, 110 mya for the split of Pelobatidae and Pelodytidae families43, 145 mya for the split of Pelobatidae and Neobatrachia43, and 61 mya for the split of Plethodidae and Proteidae44. Additionally, we set a minimum and maximum age (312-330 mya) for the split between diaspid (Gallus gallus, Xenopeltis unicolor) and synapsid amniotes (Mus musculus), based on Benton and Donoghue45.
The data matrix and the phylogenetic tree with the highest likelihood are available in Treebase (accession number: S13561).
Birds
We include here 100 dated phylogenetic trees for 430 species of European breeding birds from Roquet et al20. This phylogenetic dataset was built upon sequences retrieved from GenBank for 10 mitochondrial gene regions (12S , ATP6 , ATP8 , COII , COIII , ND1 , ND3 , ND4 , ND5 , ND6) and six nuclear ones (28S , c-mos, c-myc , RAG-1 , RAG-2 , ZENK). The alignment procedure was the same as for Amphibians. We also performed 100 ML phylogenetic inference searches and standard bootstrapping (1000 replicates) with RaxML, applying a tree constraint at the ordinal level based on Hackett et al15. The 100 trees were dated with penalized likelihood (r8s) applying fossil calibrations for 14 clades (Table S2, Appendix 2). The best ML tree can be found in Treebase (study number 10770).
Mammals
The phylogenetic data here included for mammals is based on the super-tree of Fritz and colleagues46; concretely, we extracted the resampled dataset of 100 fully resolved phylogenetic trees from Kuhn et al.47, where polytomies of the super tree from Fritz et al.46 were randomly resolved applying a birth-death model to simulate branch-lengths. Then, for each tree, we replaced the Carnivora clade with the update performed on a recent study48, which provides a better resolution and increases the sampling from 252 sp to all Carnivora species (286 sp). Later, we removed all non-European species. These modifications of the phylogenetic trees were done with the R package ape.
Phylogenetic inference
As stated before, for each taxon group except mammals we have conducted 100 ML inferences with RAxML. Every inference begins with a different starting tree, which is built by adding sequences one by one in random order, identifying their optimal location on the tree under the parsimony optimality criterion. Since sequences are added in random order, it is very likely that a different starting tree is generated at every search8788. RAxML searches were then performed with the method “lazy subtree rearrangement” (a variant of subtree prunning and regrafting method) under a ML framework. Like all heuristic search strategies, the Maximum Likelihood search strategy employed by RAxML is not guaranteed to find the most probable tree of the tree-space, and because of that, it is important to conduct multiple searches from different starting trees. To check if all the searches converged on trees with similar likelihoods, we performed the Shimodaira-Hasegawa test89 (SH) implemented in RAxML. In all cases, the likelihoods of the trees of a same group of taxa were not significantly different (p < 0.01). This increases our confidence on the trees found being close/similar to the most likely tree, and that the trees obtained do not result from the algorithm getting stuck in a local optima.
Supertree construction
The trees cited above were combined, after pruning the outgroups, by joining them with the R package ape; to do so we set divergence ages between these main groups based on the information retrieved in the webpage Timetree49: the divergence age between mammals and sauropsids (i.e. birds, turtles and squamates) was set to 324 mya, and the divergence age between amphibians and the rest of the groups was set to 361 mya. To build the final tetrapod tree, we randomly selected one tree from each of 100 trees available for each group. We repeated this approach 100 times to get 100 realisations of the tetrapod tree. These combinations were done randomly since the likelihoods of the trees of each group were not significantly different according to the SH test. The 100 dated trees of each group and the 100 dated supertrees for all European Tetrapoda are available from the Dryad digital repository (DOI: X).
Results and Discussion
Squamata
The study of Pyron et al.14 constituted a major advance in our understanding of the phylogenetic relationships between the main lineages of Squamata. Their study had a broad taxonomic and molecular sampling: they included members of all currently recognized families and subfamilies, for which 7 nuclear and 5 mitochondrial loci were analysed. Here, we took profit of the knowledge derived from that study by incorporating a tree constraint to the family level based on their results. We also performed the analysis without the tree constraint (results not shown); the results were congruent with the first analysis, but the lack of a family-tree constraint yield low bootstrap (BS) support for the deepest nodes.
Our phylogenetic results are largely congruent with those of Pyron and colleagues14. We have similar levels of strong nodal supports except for the relationships between genera of Lacertidae; 67.8% of the nodes had a strong support (BS > 70%, Fig. 1, Appendix 3) and 13.1% of the nodes had a moderate support (BS 50-70%). In accordance with their study, we detected that some genera are not monophyletic: Ablepharus (Scincidae), Cyrtopodion (Gekkonidae), Zamenis (Colubridae). We also found strong evidence that Hierophis and Dolichophis (Colubridae) are not monophyletic genera, as D. cypriensis (which was not included in 14) is nested within Hierophis with a 100% BS.
Available dating studies on Squamata differ considerably on age estimates. For instance, a recent study30 estimated the squamate crown group to be c. 180 mya, while two other studies estimated the same group to be c. 240 mya50,51. Our estimates of divergence times are in general roughly similar to those of Kumazawa’s study50. It has been suggested30 that the use of only mitochondrial regions (which is not the case here) may bias the results towards older ages, but anyway differences in methodology and in taxon and molecular sampling make difficult to identify all the causes of those discrepancies.
Amphibians
The phylogenetic inference analysis for the amphibians yielded a particularly robust topology: 83.5 % of the nodes showed a strong support (BS>70%, Fig. 2, Appendix 3). Supported nodes of our ML trees were congruent with previous phylogenetic studies52,53,54,55,56,57. Concerning the divergence age estimates, we obtained younger ages for the deepest nodes compared to the work of Roelants and colleagues13, for instance, Batrachia was estimated with r8s to be c. 330 mya in that study, while we estimated it to be c. 300 mya; in contrast, we retrieved older ages for the shallowest nodes (e.g. we estimated that the divergence between Salamandra and Pleurodeles occurred 100 mya, Roelants and colleagues estimated it to have occurred c. 75 mya). These differences might be linked to the difference in molecular and taxon sampling: Roelants et al. sampled only one species per genera; and several families that were included in their work are not present in Europe and thus were not included in our supermatrix.
Birds
Supported nodes of our ML trees are congruent with previous phylogenetic studies (Anseriformes: Donne-Goussé et al. 58, Eo et al.59, Gonzalez et al.60; Galliformes: Gutierrez et al.61 , Dimcheff et al.62, Crowe et al.63, Kimball et al.64, Kriegs et al.65, Lislevand et al.66; Gruiformes: Fain et al.67; Procellariiformes: Penhallurick and Wink68; Ardeidae: Sheldon et al.69 ; Accipitridae: Lerner and Mindell70, Griffiths et al.71 ; Charadriiformes: Paton et al.72 , Thomas et al.73, Pons et al.74 , Bridge et al.75 , Paton and Baker76 , Fain and Houde77; Passeriformes: Alström et al.78, Nguembock et al.79, Treplin et al.80; Piciformes-Coraciiformes: Johansson et al.81, Benz et al.82; Strigiformes: Wink et al.83); 68.7% of the nodes had a strong BS support (BS>70%, Fig. 3, Appendix 3), and an additional 12.4% had a moderate support (BS=50-70%). Divergence age estimates were, in general, congruent with those obtained by Brown et al84.
Mammals
The modification of the most recent mammals supertrees available on the literature47 with the update of Carnivora clade48 allowed to increase the phylogenetic resolution (only nine polytomies remain in the updated Carnivora clade) and to have a higher species sampling.
The importance of accounting for phylogenetic uncertainty
Phylogenetic information is sometimes incorporated in ecological analyses based on a single phylogenetic tree, assuming the tree is known without error. Any phylogenetic tree estimate will probably not be an exact representation of the true phylogeny due to possible bias or uncertainties such as molecular and taxon sampling, sequence alignment, homoplasy, or long-branch attraction9091. For all these reasons, it is important to include phylogenetic uncertainty in order to avoid overestimating our confidence in subsequent analyses (i.e. obtaining too narrow confidence intervals). This type of uncertainty can be accounted for in two ways: with a single consensus tree (in which unsupported nodes are collapsed into polytomies), or running the analyses with a range of trees and later summarising the results11, 93. The first approach (i.e. consensus tree) may not be preferable, as polytomies can influence the results of tree-based statistical analyses (e.g. see 92 for the influence of phylogenetic resolution on several community phylogenetics indices), and do not allow to fully explore the variation in ecological patterns resulting from phylogenetic uncertainty. Moreover, not all methods have been adapted to allow for polytomies, some of them require completely bifurcating trees (e.g. the EDGE index10). For all these reasons, we highly recommend to account for phylogenetic uncertainty by including a set of high-probability trees.
Data availability statement
The 100 dated supertrees for all European Tetrapoda and the 100 dated trees of each taxon group (amphibians, birds, mammals, squamates and turtles) are available from the Dryad digital repository (DOI: X).
Conclusion
We provide here a phylogenetic dataset constituted of 100 chronograms of European Tetrapoda species as a tool for ecological studies that aim to incorporate an evolutionary perspective, and for phylogenetic conservation assessment. This phylogenetic dataset is in general agreement with previous studies, and we expect it to be coarsely approximate with the “true” Tetrapoda evolutionary tree. Instead of providing the best ML tree for every group, we provide 100 trees (available on Dryad repository), as computing analyses with several trees allows taking in account phylogenetic uncertainty. Regarding the taxonomic sampling, the big majority of species are included (91%). On the other side, some molecular regions have low sampling, thus, this dataset will be useful until substantial amount of molecular data becomes available for a considerable number of species.
Competing Interests
The authors have declared that no competing interests exist.
Appendix 1
Genbank accession numbers for squamates and turtles
Appendix 2
Calibration of the bird phylogenetic trees
Table S2. Summary of the fossil data used for calibration of the phylogenetic trees of birds. All fossils were used as minimum age constraints, except *, which was used as a maximum age.
Clade
Age
Stem/Crown
References
Anseriformes
66
Stem
Clarke et al. (2005)
Ingroup
125*
Stem
Zhou (2004)
Apodiformes
53
Stem
Mayr (2003)
Upupidae+ Picidae
47.05.00
Stem
Mayr (2000)
Coraciidae+ Meropidae
47.05.00
Stem
Mayr & Mourer-Chauviré (2000)
Pandionidae
37
Crown
Harrison & Walker (1976)
Sulidae
33
Stem
Mayr (2002)
Pteroclidae
30
Stem
Mourer-Chauviré (1993)
Rallidae
33
Stem
Mayr & Smith (2001)
Scolopaciade
33
Stem
Olson (1985)
Strigidae
58
Stem
Vickers-Rich & Bohaska (1976)
Procellariidae
23
Stem
Olson (1985)
Sulidae
33
Stem
Mayr (2002)
Burhinidae
18
Stem
Bickart (1982)
Additional references
Clarke JA, Tambussi CP, Noriega JI, Erickson GM, Ketcham RA. 2005. Definitive fossil evidence for the extant avian radiation in the Cretaceous. Nature 433: 305-308.
Harrison CJO, Walker CA. 1976. Birds of the British Upper Eocene. Zoological Journal of the Linnean Society 59: 323-351.
Mayr G. 2000. Tiny hoopoe-like birds from the Middle Eocene of Messel (Germany). Auk 117: 968-974.
Mayr G. 2002. A skull of a new pelecaniform bird from the Middle Eocene of Messel, Germany. Acta Palaeontologica Polonica 47: 507-512.
Mayr G. 2003. Phylogeny of early Tertiary swifts and hummingbirds (Aves: Apodiformes). Auk 120: 145-151.
Mayr G, Mourer-Chauviré C. 2000. Rollers (Aves: Coraciiformes s.s.) from the Middle Eocene of Messel (Germany) and the Upper Eocene of the Quercy (France). Journal of Vertebrate Paleontology 20: 533-546.
Mayr G, Smith R. 2001. Duck, rails, and limicoline waders from the lowermost Oligocene of Belgium. Geobios 34: 547–561.
Mourer-Chauviré C. 1993. Les gangas (Aves, Columbiformes, Pteroclidae) du Paléogène et du Miocène inférieur de France. Palaeovertebrata 22: 73-98.
Olson SL. 1985. The fossil record of birds. In: Farner D (ed.), Avian Biology. Academic Press, pp. 80–238.
Vickers-Rich P, Bohaska DJ. 1976. The world’s oldest owl: A new 1944 strigiform from the Paleocene of Southwestern Colorado. 1945 Smithsonian Contributions to Paleobiology 27:87–93.
Zhou Z. 2004. The origin and early evolution of birds: discoveries, disputes, and perspectives from fossil evidence. Naturwissenschaften 91: 455-471.
Appendix 3
Phylogenetic Trees
Phylogenetic tree obtained by maximum likelihood inference with RaxML (outgroups have been removed). Numbers above branches indicate bootstrap values.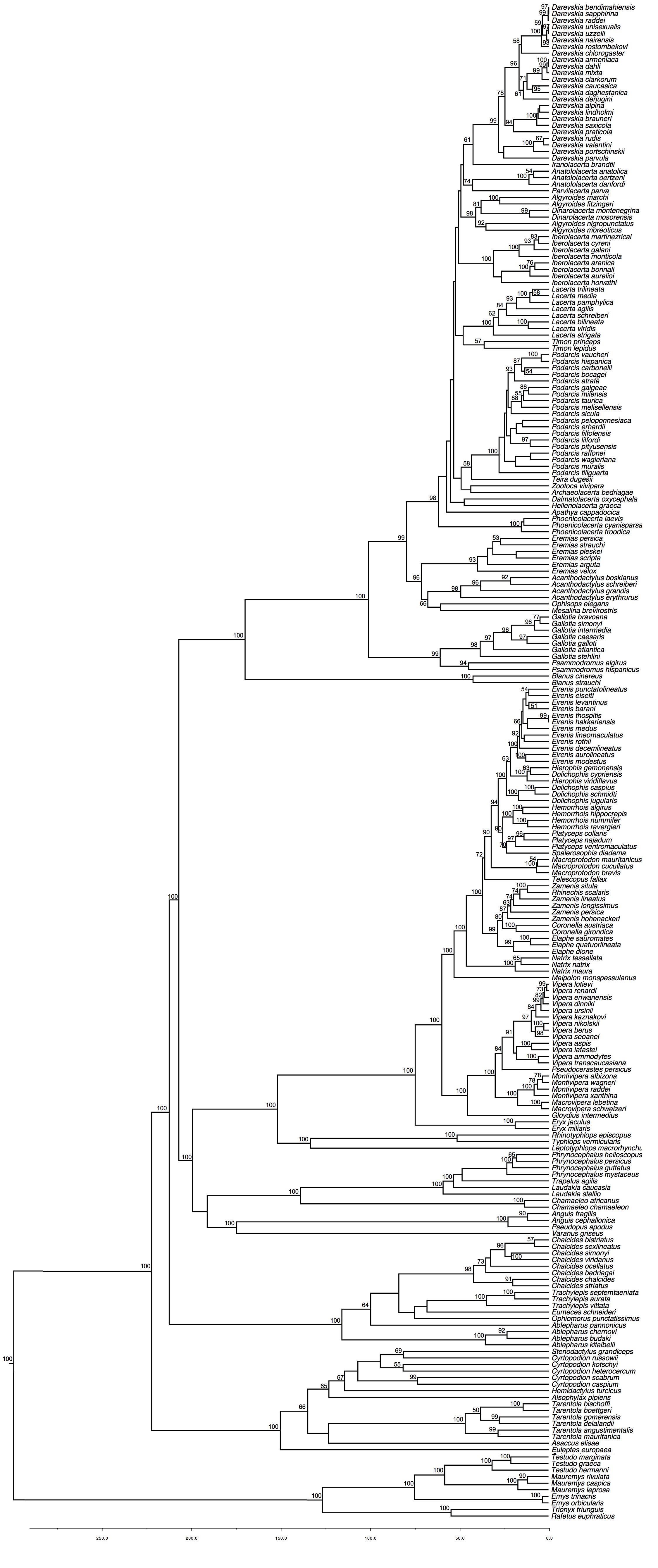
Fig. 1: Phylogenetic tree of European squamates and turtles
Phylogenetic tree obtained by maximum likelihood inference with RaxML (outgroups have been removed). Numbers above branches indicate bootstrap values.
Fig. 2: Phylogenetic tree of European amphibians
Phylogenetic tree obtained by maximum likelihood inference with RaxML. Numbers above branches indicate bootstrap values.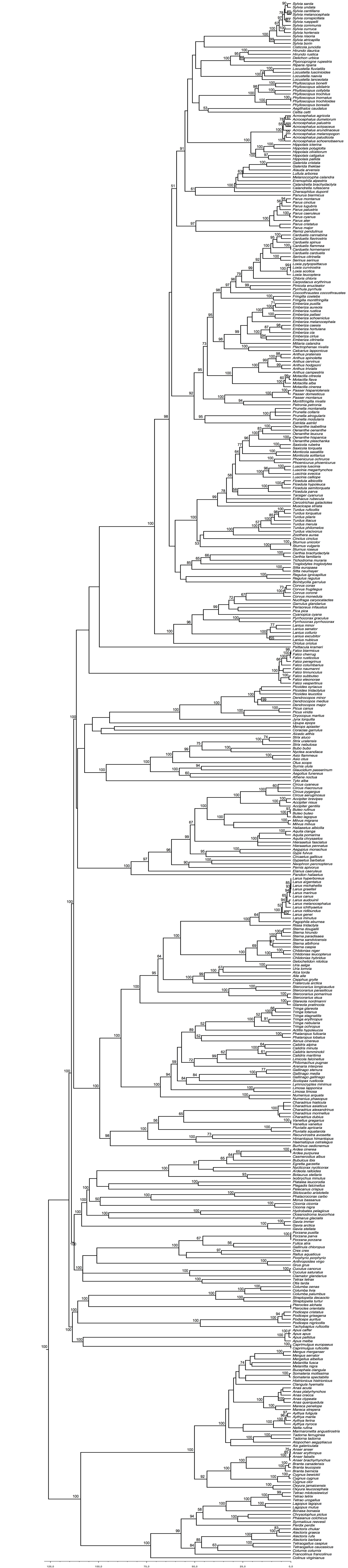
Fig. 3: Phylogenetic tree of European birds
References
- Webb CO, Ackerly DD, McPeek MA, Donogue MJ. 2002. Phylogenies and community ecology. Annual Review of Ecology and Systematics 33: 475–505.
- Mouquet N, Devictor V, Meynard CN, Munoz F, Bersier L-F, Chave J, Couteron P, Dalecky A, Fontaine C, Gravel D, Hardy OJ, Jabot F, Lavergne S, Leibold M, Mouillot D, Münkemüller T, Pavoine S, Prinzing A, Rodrigues ASL, Rohr RP, Thébaut E, Thuiller W. 2012. Ecophylogenetics: advances and perspectives. Biological Reviews 87: 769–785.
- Jetz W, Thomas GH, Joy JB, Hartmann K, Mooers AO. 2012. The global diversity of birds in space and time. Nature 491: 444–448.
- Davies TJ, Buckley LB. 2012. Exploring the phylogenetic history of contemporary mammal species richness. Global Ecology and Biogeography 21: 1096–1105.
- Lavergne S, Mouquet N, Thuiller W, Ronce O. 2010. Biodiversity and climate change: integrating evolutionary and ecological responses of species and communities. Annual Review of Ecology, Evolution and Systematics 41: 321–350.
- Thuiller W, Münkemüller T, Lavergne S, Mouillot D, Mouquet N, Schiffers K, Gravel D. 2013. A road map for integrating eco-evolutionary processes into biodiversity models. 2013. Ecology Letters 16: 94–105.
- Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biological Conservation 61: 1–10.
- Mace GM, Gittleman JL, Purvis A. 2003. Preserving the Tree of Life. Science 300: 1707–1709.
- Cadotte MW, Davies TJ, Regetz J, Kembel SW, Cleland E, Oakley TH. 2010. Phylogenetic diversity metrics for ecological communities: integrating species richness, abundance and evolutionary history. Ecology Letters 13: 96–105.
- Isaac NJB, Turvey ST, Collen B, Waterman C, Baillie JEM. 2007. Mammals on the EDGE: conservation priorities based on threat and phylogeny. PLoS ONE 2: e296.
- Thuilller W, Lavergne S, Roquet C, Boulangeat I, Lafourcade B, Araújo MB. 2011. Consequences of climate change on the tree of life in Europe. Nature 470: 531–534.
- Faith D, Richards ZT. 2012. Climate change impacts on the Tree of Life: changes in phylogenetic diversity illustrated for Acropora corals. Biology 1: 906–932.
- Roelants K, Gower DJ, Wilkinson M, Loader SP, Biju SD, Guillaume K, Moriau L, Bossuyt F. 2007. Global patterns of diversification in the history of modern amphibians. Proceedings of the National Academy of Sciences of the United States of America 104: 887–92.
- Pyron RA, Burbrink FT, Wiens JJ. 2013. A phylogeny and revised classification of Squamata, incluidng 4161 species of lizards and snakes. BMC Evolutionary Biology 13: 93.
- Hackett SJ, Kimball RT, Reddy S, Bowie RCK, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han KL, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Sheldon FH, Steadman DW, Witt CC, Yuri T. 2008. A phylogenomic study of birds reveals their evolutionary history. Science 320: 1763–1768.
- McCormack JE, Harvey MG, Faircloth BC, Crawford NG, Glenn TC, Brumfield RB. 2013. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS One 8: e54848.
- Durka W, Michalski SG. 2012. Daphne: a dated phylogeny of a large European flora for phylogenetically informed ecological analyses. Ecology 93: 2297–2297.
- Münkemüller T, Gallien L, Lavergne S, Renaud J, Roquet C, Abdulhak S, Dullinger S, Garraud L, Guisan A, Lenoir J, Svenning J-C, Van Es J, Willner W, Wohlgemuth T, Zimmermann N, Thuiller W. 2014. Scale decisions can reverse conclusions on community assembly processes. Global Ecology and Biogeography 23: 620–632.
- Zupan L, Cabeza M, Maiorano L, Roquet C, Devictor V, Lavergne S, Mouillot D, Mouquet N, Renaud J, Thuiller W. In press. Spatial mismatch of phylogenetic diversity across three vertebrate groups and protected areas in Europe. Diversity and Distributions. DOI: 10.1111/ddi.12186
- Roquet C, Thuiller W, Lavergne S. 2013. Building megaphylogenies for macroecology: taking up the challenge. Ecography 36: 13–26.
- Maiorano L, Amori G, Capula M, Falcucci A, Masi M, Montemaggiori A, Pottier J, Psomas A, Rondinini C, Russo D, Zimmermann NE, Boitani L, Guisan A. 2013. Threats from climate change to terrestrial vertebrate hotspots in Europe. PLoS ONE 8: e74989.
- Smith SA, Beaulieu J, Donoghue MJ. 2009. Mega-phylogeny approach for comparative biology: an alternative to supertree and supermatrix approaches. BMC Evol Biol 9: 37.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780.
- Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4 : a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution 27: 221–224.
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–1973.
- Kück P, Meusemann K. 2010. FASconCAT Version 1.0, Zool. Forschungsmuseum A. Koenig, Germany.
- Stamatakis A, Hoover P, Rougemont J. 2008. A fast bootstrapping algorithm for the RAxML Web-Servers. Systematic Biology 57: 758–771.
- Stamatakis A, Blagojevic F, Nikolopoulos D, Antonopoulos C. 2007. Exploring new search algorithms and hardware for phylogenetics: RAxML meets the IBM cell. Journal of VLSI Signal Processing Systems 48: 271–286.
- Sanderson MJ. 2003. r8s: inferring absolute rates of evolution and divergence times in the absence of a molecular clock. Bioinformatics 19: 301–302.
- Mulcahy DG, Noonan BP, Moss T, Townsend TM, Reeder TW, Sites JW Jr, Wiens JJ. 2012. Estimating divergence dates and evaluating dating methods using phylogenomic and mitochondrial data in squamate reptiles. Molecular Phylogenetics and Evolution 65: 974–991.
- Donoghue PCJ, Benton MJ. 2007. Rocks and clocks: calibrating the tree of life using fossils and molecules. Trends in Ecology & Evolution 22: 424–431.
- Benton MJ, Donoghue PCJ, Asher RJ. 2009. Calibrating and constraining the molecular clock. In: Hedges SB, Kumar S (eds.), The Timetree of Life. Oxford University Press, pp. 35–86.
- Sues HD, Olsen PE. 1990. Triassic vertebrates of Gondwanan aspect from the Richmond Basin of Virginia. Science 249: 1020–1023.
- Evans SE. 2003. At the feet of the dinosaurs: the early history and radiation of lizards. Biological Reviews 78: 513–551.
- Wiens JJ, Brandley MC, Reeder TW. 2006. Why does a trait evolve multiple times within a clade? Repeated evolution of snakelike body form in squamate reptiles. Evolution 60: 123–141.
- Marsh OC. 1892. Notice of new reptiles from the Laramie Formation. American Journal of Science 43: 449–453.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948.
- Lassmann T, Sonnhammer ELL. 2005 Kalign – an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics 6: 298.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797.
- Evans SE, Lally C, Chure DC, Elder A, Maisano JA. 2005. A Late Jurassic salamander (Amphibia: Caudata) from the Morrison Formation of North America. Zoological Journal of the Linnean Society 143: 599–616.
- Evans SE, Milner AR, Musset F. 1990. A discoglossid frog from the Middle Jurassic of England. Palaeontology 33: 299–311.
- Rage J-C, Roček Z. 1989. Redescription of Triadobatrachus massinoti (Piveteau,1936) an anuran amphibian from the Early Triassic. Palaeontographica Abstract A 206: 1–16.
- Evans SE, Milner AR. 1993. Frogs and salamanders from the Upper Jurassic Morrison Formation (Quarry Nine, Como Bluff) of North America. Journal of Vertebrate Paleontology 13: 24–30.
- Gardner JD. 2003 The fossil salamander Proamphiuma cretacea Estes (Caudata; Amphiumidae) and relationships within the Amphiumidae. Journal of Vertebrate Paleontology 23: 769–782.
- Benton MJ, Donoghue PCJ. 2007. Paleontological evidence to date the tree of life. Molecular Biology and Evolution 24: 26–53.
- Fritz SA, Bininda-Emonds ORP, Purvis A. 2009. Geographical variation in predictors of mammalian extinction risk: big is bad, but only in the tropics. Ecology Letters 12: 538–549.
- Kuhn TS, Mooers AØ, Thomas GH. 2011. A simple polytomy resolver for dated phylogenies. Methods in Ecology and Evolution 2: 427–436.
- Nyakatura K, Bininda-Emonds ORP. 2012. Updating the evolutionary history of Carnivora (Mammalia): a new species-level supertree complete with divergence time estimates. BMC Biology 10: 12.
- Hedges SB, Dudley J, Kumar S. 2006. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22: 2971–2972.
- Kumazawa Y. 2007. Mitochondrial genomes from major lizard families suggest their phylogenetic relationships and ancient radiations. Gene 388: 19–26.
- Vidal N, Hedges SB. 2005. The phylogeny of squamate reptiles (lizards, snakes and amphisbaenians) inferrred from nine nuclear protein-coding genes. Comptes Rendus Biologies 328: 1000–1008.
- Veith M, Kosuch J, Vences M. 2003. Climatic oscillations triggered post-Messinian speciation of Western Palearctic brown frogs (Amphibia, Ranidae). Molecular Phylogenetics and Evolution 26: 310–327.
- Van der Meijden A, Chiari Y, Mucedda M, Carranza S, Corti C, Veith M. 2009. Phylogenetic relationships of Sardinian cave salamanders, genus Hydromantes,based on mitochondrial and nuclear DNA sequence data. Molecular Phylogenetics and Evolution 51: 399–404.
- Zhang P, Papenfuss TJ, Wake MH, Qu LH, Wake DB. 2008. Phylogeny and biogeography of the family Salamandridae (Amphibia: Caudata) inferred from complete mitochondrial genomes. Molecular Phylogenetics and Evolution 49: 586–597.
- Stöck M, Sicilia A, Belfiore NM, Buckley D, Lo Brutto S, Lo Valvo M, Arculeo M. 2008. Post-Messinian evolutionary relationships across the Sicilian channel: Mitochondrial and nuclear markers link a new green toad from Sicily to African relatives. BMC Evolutionary Biology 8: 56.
- Hua X, Fu C, Li J, Montes de Oca AN, Wiens JJ. 2009 A Revised Phylogeny of Holarctic Treefrogs (Genus Hyla) based on Nuclear and Mitochondrial DNA Sequences. Herpetologica 65: 246–259.
- Wiens JJ, Sparreboom M, Arntzen JW. 2011. Crest evolution in newts: implications for reconstruction methods, sexual selection, phenotypic plasticity and the origin of novelties. Journal of Evolutionary Biology 24: 2073–2086.
- Donne-Goussé C, Laudet V, Hänni C. 2002. A molecular phylogeny of anseriformes based on mitochondrial DNA analysis. Molecular Phylogenetics and Evolution 23: 339–356.
- Eo SH, Bininda-Emonds ORP, Carroll JP. 2009. A phylogenetic supertree of the fowls (Galloanserae, Aves). Zoologica Scripta 38: 465–481.
- Gonzalez J, Düttmann H, Wink M. 2009. Phylogenetic relationships based on two mitochondrial genes and hybridization patterns in Anatidae. Journal of Zoology 279: 310–318.
- Gutierrez RJ, Barrowclough GF, Groth JG. 2000. A classification of the grouse (Aves : Tetraoninae) based on mitochondrial DNA sequences. Wildlife Biology 6: 205–211.
- Dimcheff DE, Drovetski SV, Mindell DP. 2002. Phylogeny of Tetraoninae and other galliform birds using mitochondrial 12S and ND2 genes. Molecular Phylogenetics and Evolution 24: 203–215.
- Crowe TM, Bowie RCK, Bloomer P, Mandiwana T, Hedderson T, Randi E, Pereira SL, Wakeling J. 2006. Phylogenetics and biogeography of, and character evolution in gamebirds (Aves: Galliformes): effects of character exclusion, partitioning and missing data. Cladistics 22: 1–38.
- Kimball RT, Braun EL, Ligon JD, Randi E, Lucchini V. 2006. Using molecular phylogenetics to interpret evolutionary changes in morphology and behavior in the Phasianidae. Acta Zoologica Sinica 52 (supplement): 362–365.
- Kriegs JO, Matzke A, Churakov G, Kuritzin A, Mayr G, Brosius J, Schmitz J. 2007. Waves of genomic hitchhikers shed light on the evolution of gamebirds (Aves: Galliformes). BMC Evolutionary Biology 7: 190.
- Lislevand T, Figuerola J, Székely T. 2009. Evolution of sexual size dimorphism in grouse and allies (Aves: Phasianidae) in relation to mating competition, fecundity demands and resource division. Journal of Evolutionary Biology 22: 1895–1905.
- Fain MG, Krajewski C, Houde P. 2007. Phylogeny of "core Gruiformes" (Aves: Grues) and resolution of the Limpkin-Sungrebe problem. Molecular Phylogenetics and Evolution 43: 515–529.
- Penhallurick J, Wink M. 2004. Analysis of the taxonomy and nomenclature of the Procellariformes based on complete nucleotide sequences of the mitochondrial cytochrome b gene. Emu 104: 125–147.
- Sheldon FH, Jones CE, McCracken KG. 2000. Relative patterns and rates of evolution in heron nuclear and mitochondrial DNA. Molecular Biology and Evolution 17: 437–450.
- Lerner HRL, Mindell DP. 2005. Phylogeny of eagles, Old World vultures, and other Accipitridae based on nuclear and mitochondrial DNA. Molecular Phylogenetics and Evolution 37: 327–346.
- Griffiths CS, Barrowclough GF, Groth JG, Mertz LA. 2007. Phylogeny, diversity, and classification of the Accipitridae based on DNA sequences of the RAG-1 exon. Journal of Avian Biology 38: 587–602.
- Paton TA, Baker AJ, Groth JG, Barrowclough GF. 2003. RAG-1 sequences resolve phylogenetic relationships within charadriiform birds. Molecular Phylogenetics and Evolution 29: 268–278.
- Thomas GH, Wills MA, Székely T. 2004. A supertree approach to shorebird phylogeny. BMC Evolutionary Biology 4: 28.
- Pons J-M, Hassanin A, Crochet P-A. 2005. Phylogenetic relationships within the Laridae (Charadriiformes: Aves) inferred from mitochondrial markers. Molecular Phylogenetics and Evolution 37: 686–699.
- Bridge ES, Jones AW, Baker AJ. 2005. A phylogenetic framework for the terns (Sternini) inferred from mtDNA sequences: implications for taxonomy and plumage evolution. Molecular Phylogenetics and Evolution 35: 459–469.
- Paton TA, Baker AJ. 2006. Sequences from 14 mitochondrial genes provide a well-supported phylogeny of the Charadriiform birds congruent with the nuclear RAG-1 tree. Molecular Phylogenetics and Evolution 39: 657–667.
- Fain MG, Houde P. 2007. Multilocus perspectives on the monophyly and phylogeny of the order Charadriiformes (Aves). BMC Evolutionary Biology 7: 35.
- Alström P, Olsson U, Lei F, Wang HT, Gao W, Sundberg P. 2008. Phylogeny and classification of the Old World Emberizini (Aves, Passeriformes). Molecular Phylogenetics and Evolution 47: 960–973.
- Nguembock B, Fjeldså J, Couloux A, Pasquet E. 2008. Phylogeny of Laniarius: molecular data reveal L. liberatus synonymous with L. erlangeri and "plumage coloration" as unreliable morphological characters for defining species and species groups. Molecular Phylogenetics and Evolution 48: 396–407.
- Treplin S, Siegert R, Bleidorn C, Thompson HS, Fotso R, Tiedemann R. 2008. Molecular phylogeny of songbirds (Aves: Passeriformes) and the relative utility of common nuclear marker loci. Cladistics 24: 328–349.
- Johansson US, Parsons TJ, Irestedt M, Ericson PGP. 2001. Clades within the `higher land birds', evaluated by nuclear DNA sequences. Journal of Zoological Systematics and Evolutionary Research 39: 37–51.
- Benz BW, Robbins MB, Peterson AT. 2006. Evolutionary history of woodpeckers and allies (Aves: Picidae): Placing key taxa on the phylogenetic tree. Molecular Phylogenetics and Evolution 40: 389–399.
- Wink M, El-Sayed AA, Sauer-Gürth H, Gonzalez J. 2009. Molecular phylogeny of owls (Strigiformes) inferred from DNA sequences of the mitochondrial cytochrome b and the nuclear RAG-1 gene. Ardea 97: 581–591.
- Brown JW, Rest JS, García-Moreno J, Sorenson MD, Mindell DP. 2008. Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biology 6: 6.
- Devictor V, Mouillot D, Meynard C, Jiguet F, Thuiller W, Mouquet N. Spatial mismatch and congruence between taxonomic, phylogenetic and functional diversity: the need for integrative conservation strategies in a changing world. Ecol Lett. 2010 Aug 1;13(8):1030-40. PubMed PMID:20545736.
- Thuiller W, Maiorano L, Mazel F, Guilhaumon F, Ficetola GF, Lavergne S, Renaud J, Roquet C, Mouillot D. In press. Conserving the functional and phylogenetic trees of life of European tetrapods. Philosophical Transactions of the Royal Society B: Biological Sciences.
- Stamatakis A, Ludwig T, Meier H. 2005. RAXML-III: A fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21: 456-463.
- Rokas A. 2011. Phylogenetic analysis of protein ssequence data using the randomized axelerated maximum likelihood (RAxML) program. Current Protocols in Molecular Biology, 19.11.1-19.11.14
- Shimodaira H, Hasegawa M. 1999. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution 16: 1114–1116.
- Harvey PH, Pagel MD. 1991. The Comparative Method in Evolutionary Biology. Oxford, Oxford University Press.
- Donoghue MJ, Ackerly DD. 1996. Phylogenetic Uncertainties and Sensitivity Analyses in Comparative Biology. Philosophical Transactions of the Royal Society B: Biological Sciences 351: 1241–1249.
- Molina-Venegas R, Roquet C. 2014. Directional biases in phylogenetic structure quantification: a Mediterranean case study. Ecography, 37: 572-580.
- Lavergne S, Evans MEK, Burfield IJ, Jiguet F, Thuiller W 2013. Are species’ responses to global change predicted by past niche evolution? Philosophical Transactions of the Royal Society B: Biological Sciences 368: 20120091.
Cristina Roquet
The identifier of the tree datasets included in Dryad is: doi:10.5061/dryad.11609