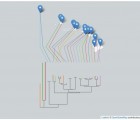
Orchidaceae constitutes one of the largest families of angiosperms. Owing to the significance of orchids in plant biology, market needs and current sustainable technology levels, basic research on the biology of orchids and their applications in the orchid industry is increasing. Although chloroplast (cp) genomes continue to be evolutionarily informative, there is very limited information available on orchid chloroplast genomes in public repositories. Here, we report the complete cp genome sequence of Dendrobium nobile from Northeast India (Orchidaceae, Asparagales), bearing the GenBank accession number KX377961, which will provide valuable information for future research on orchid genomics and evolution, as well as the medicinal value of orchids. Phylogenetic analyses using Bayesian methods recovered a monophyletic grouping of all Dendrobium species (D. nobile, D. huoshanense, D. officinale, D. pendulum, D. strongylanthum and D. chrysotoxum). The relationships recovered among the representative orchid species from the four subfamilies, i.e., Cypripedioideae, Epidendroideae, Orchidoideae and Vanilloideae, were consistent within the family Orchidaceae.